KIMMDY
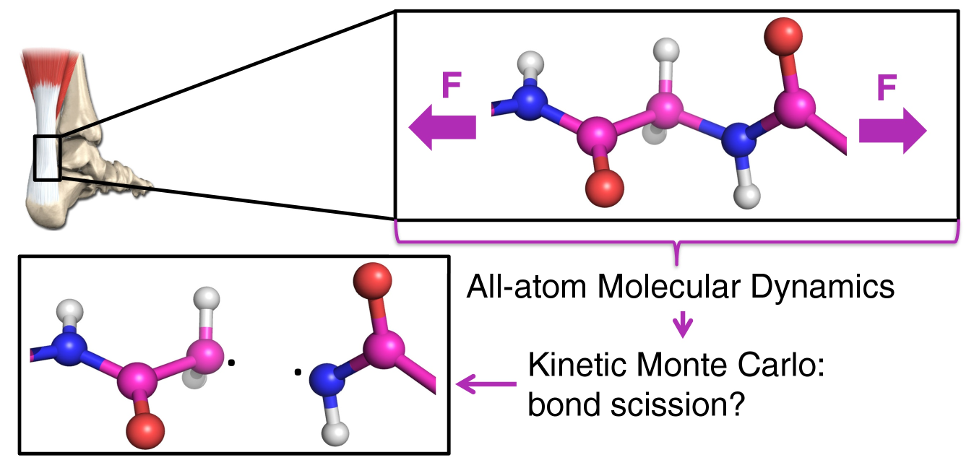
We have developed a simulation scheme called KIMMDY (Kinetic Monte Carlo / Molecular Dynamics) that enables covalent bond breakages in all-atom Molecular Dynamics (MD) simulations. The bond rupture rates are calculated based on the interatomic distances in the MD simulation and then serve as an input for a Kinetic Monte Carlo step. This hybrid approach allows to study breakages in large molecular systems (like the structural protein collagen) bridging various time scales between MD and the rupture processes.
Read the detailed description of the method here.
Hybrid Kinetic Monte Carlo / Molecular Dynamics Simulations of Bond Scissions in Proteins
Benedikt Rennekamp, Fabian Kutzki, Agnieszka Obarska-Kosinska, Christopher Zapp, and Frauke Gräter
Journal of Chemical Theory and Computation
The code and some example files can be found on GitHub.