Proteinanalyse: Weniger ist mehr
CONAN hilft! Das neue Software-Programm für Molekularbiologie-Simulationen komprimiert 3D-Visualisierungen zu sogenannten „contact maps“ und erleichtert damit die Untersuchung von Proteinstrukturen. Das von den HITS Wissenschaftlern und Wissenschaftlerinnen entwickelte Tool CONAN (CONtact ANalysis) wurde nun im „Biophysical Journal“ vorgestellt.
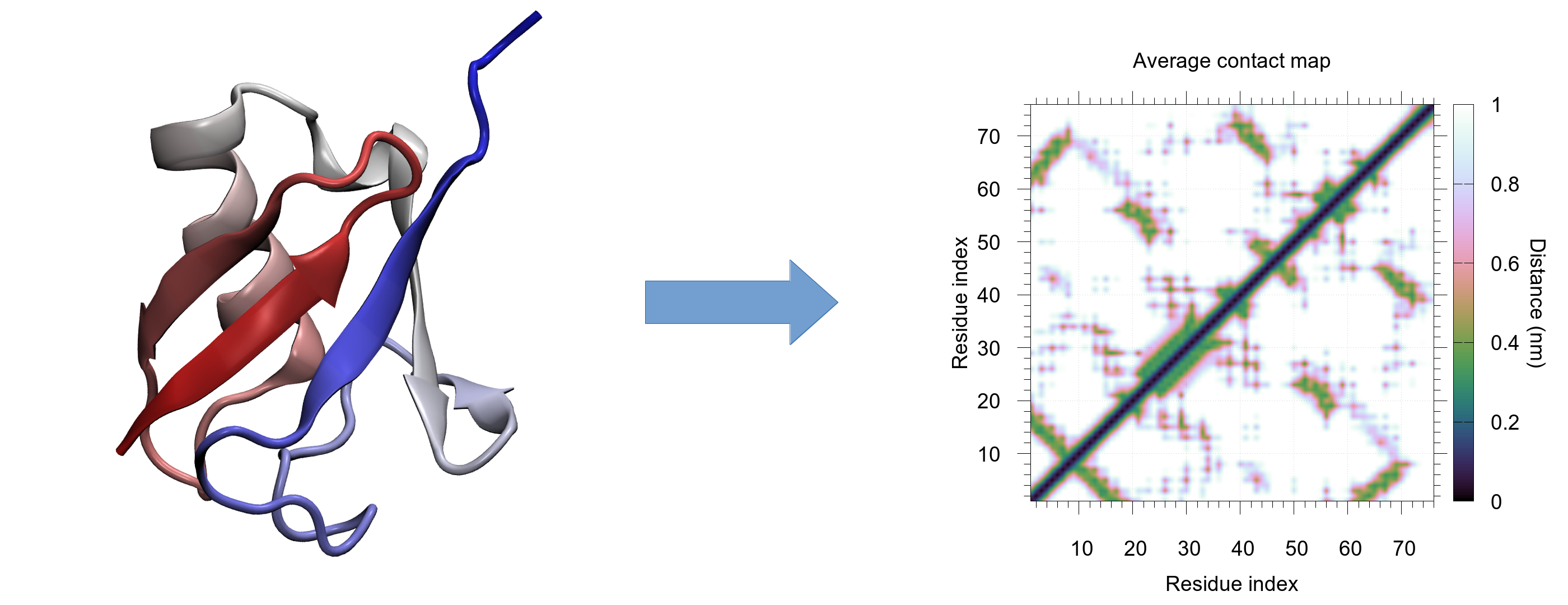
Proteine sind immer in Bewegung und wechseln dadurch ständig ihre Gestalt, auch „Konformationen“ genannt. Molekulardynamik-Simulationen beantworten typischerweise die Frage, wie mögliche Konformationen genau aussehen könnten. Die Struktur von Proteinen ist allerdings sehr komplex und gedrängt, so dass mögliche Änderungen in der Anordnung der Strukturen aufgrund der zahlreichen zu beachtenden Koordinaten eine besondere Herausforderung darstellen. Zur Verarbeitung dieser großen Datenmengen werden daher oft kreative 3D-Visualisierungen erstellt. Doch auch deren Auswertung ist sehr aufwendig und es besteht die Gefahr, dass wichtige Details übersehen werden. Dies führte bisher zu gleich zwei Problemen: Wissenschaftler hatten nicht nur Schwierigkeiten, ihre Ergebnisse zu visualisieren, es bestand für sie zudem das Risiko, wichtige Aspekte ihrer eigenen Daten zu übersehen. Das neue Analyse-Tool CONAN (CONtact ANalysis), das in der Molecular Biomechanics-Gruppe am HITS entwickelt wurde, kann diese Probleme lösen. Die Software komprimiert dazu die zuvor erstellten 3D-Visualisierungen in einfachere 2D-Bilder, bekannt als “Contact maps”, die die wichtigsten Interaktionen von Proteinen erfassen.
“Contact maps” vermessen die Abstände zwischen den Aminosäuren und fassen dadurch die 3D-Strukturen von Proteinen in 2D-Bilder zusammen. Diese Art der Darstellung vereinfacht die Interpretation der Daten, da wichtige Änderungen einfacher zu sehen sind. Bisher wurden diese “Contact maps” ausschließlich dazu genutzt eine einzige Struktur zu analysieren, also zum Beispiel zur Untersuchung eines Proteins zu einem bestimmten Zeitpunkt, wie etwa in der „Protein Data Base“ (PDB). Mit dem neuen Tool kann aber eine ganze Serie an Strukturen analysiert und sogar Animationen aus mehreren „Contact maps“ erstellt werden. Diese neue Analyse erweitert die bekannte Aussage „ein Bild sagt mehr als tausend Worte”: Statt nur ein Bild, kann nun eine ganze Bildreihe analysiert und dadurch eventuelle Untergruppen oder Übergänge in den Konformationen gefunden werden.
Bisherige Versuche die dynamische Komponente von Proteinstrukturen zu untersuchen, verliefen bisher meistens „ad hoc“ durch eigens dafür erstellte Skripte, da es schlicht kein vereinheitlichtes Tool für solche Analysen gab. Das führte dazu, dass die durchgeführten Messungen inkonsistent und miteinander nicht direkt vergleichbar waren. Die neue Software CONAN hingegen ist standardisiert, benutzerfreundlich, und bietet vielfache Analysemethoden, wie etwa Hauptkomponentenanalyse und Clusteranalyse. Damit füllt das von den HITS Wissenschaftlern Csaba Daday und Frauke Gräter, sowie HITS-Alumnus Davide Mercadante, entwickelte Tool eine Lücke. Das Software-Paket ist einfach zu nutzen und erfordert keine Erfahrung in Programmierung. Es dient dazu Wissenschaftlern und Wissenschaftlerinnen zu helfen, die sich mit Molekulardynamik-Simulationen beschäftigen, ihre Ergebnisse besser zu verstehen und zu präsentieren. Die einheitliche Verwendung dieses Tools kann dabei helfen, dass gleiche Methoden genutzt werden und damit die Ergebnisse einfacher zu vergleichen sind. Das Tool ist frei zugänglich, kostenlos und wird vom Team am HITS ständig optimiert. Bei offenen Fragen oder Feedback bitte an Csaba Daday von der Molecular Biomechanics-Gruppe wenden.
CONAN ist frei verfügbar hier.
Beispiele zur Anwendung gibt es auf dem Blog sowie auf YouTube.
Artikel im „Biophysical Journal“:
CONAN: A Tool to Decode Dynamical Information from Molecular Interaction Maps. Davide Mercadante, Frauke Gräter, Csaba Daday. Biophysical Journal,
Volume 114, Issue 6, p1267–1273, 27 March 2018. DOI: https://doi.org/10.1016/j.bpj.2018.01.033
Wissenschaftlicher Kontakt:
Prof. Dr. Frauke Gräter
Gruppenleiterin „Molecular Biomechanics“
HITS – Heidelberger Institut für Theoretische Studien
E-mail: frauke.graeter@h-its.org
Dr. Csaba Daday
Mitglied der Gruppe „Molecular Biomechanics“
HITS – Heidelberger Institut für Theoretische Studien
E-mail: Csaba.Daday@h-its.org
Über das HITS
Das HITS (Heidelberger Institut für Theoretische Studien) wurde 2010 von dem Physiker und SAP-Mitbegründer Klaus Tschira (1940-2015) und der Klaus Tschira Stiftung als privates, gemeinnütziges Forschungsinstitut gegründet. Es betreibt Grundlagenforschung in den Naturwissenschaften, der Mathematik und der Informatik. Zu den Hauptforschungsrichtungen zählen komplexe Simulationen auf verschiedenen Skalen, Datenwissenschaft und -analyse sowie die Entwicklung rechnergestützter Tools für die Forschung. Die Anwendungsfelder reichen von der Molekularbiologie bis zur Astrophysik. Ein wesentliches Merkmal des Instituts ist die Interdisziplinarität, die in zahlreichen gruppen- und disziplinübergreifenden Projekten umgesetzt wird. Die Grundfinanzierung des HITS wird von der Klaus Tschira Stiftung bereitgestellt.