Rescuing Proteins from Collapse
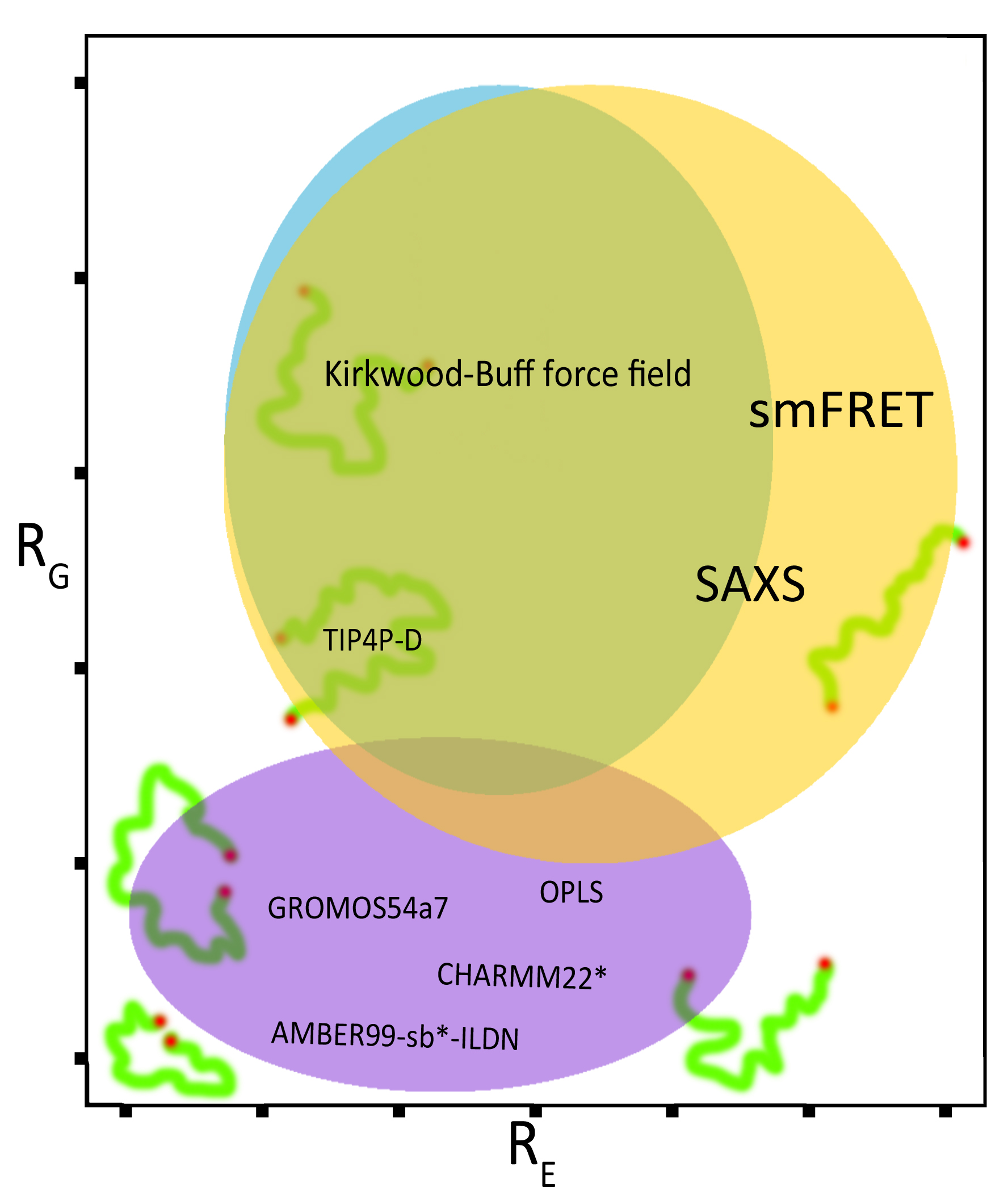
One of the cornerstones of structural biology envisions that the structure of a molecule is tightly related to its function. The recent discovery of intrinsically disordered proteins (IDPs) appeared to invalidate such a concept, as IDPs do not show any clear structure but are still able to perform complex functions inside cells. Nevertheless, IDPs tend to prefer structural arrangements along the ensemble of fluctuating conformations that they explore in solution. These conformations are the ones most likely to accomplish a function. It is therefore crucial to reproduce them in simulations. Unfortunately, one of the biggest problems of computational structural biology is that simulations of IDPs yield conformations that are simply too compact, when simulated using canonical force fields that employ parameters not developed to simulate proteins with no clear structure. A force field is a set of parameters and equations used to compute the interaction between the atoms composing the simulated molecules. Therefore they represent the barebones of Molecular Dynamics simulations. Computational structural biologists are thus unable to identify the correct mode of action of intrinsically disordered proteins. Indeed they are incapable of matching their results with observations from experiments, which report the existence of much more extended conformations. To solve this problem we have tested the ability of canonically used force fields to reproduce the correct dimensions of the conformations explored by an intrinsically disordered protein. In collaboration with experimental researchers at EMBL in Heidelberg they have assessed the extent of over-collapse that force fields produce when simulating such proteins. Most importantly, we have found that a new force field (KBFF) developed on the basis of the Kirkwood-Buff theory of solutions solves the problem of unnatural over-compaction. The Kirkwood-Buff theory of solutions links the microscopic details of the solution (intended as distribution of solvent molecules around a solute) to the macroscopic bulk properties of such a solution. It is therefore possible to obtain force field parameters directly from thermodynamic bulk, measurements. Thus, the advantage of the KBFF lies in the fact that its parameters are directly retrieved from experimental measurements. The details of their findings have been recently published in J. Phys. Chem. B (Mercadante et al.
Kirkwood-Buff Approach Rescues Overcollapse of a Disordered Protein in Canonical Protein Force Fields.
J Phys Chem B (2015) 119(25):7975-84). Now we are extending their study in collaboration with the developers of the KBFF at Kansas University, hoping to successfully test the force field on structured proteins and to validate it with new experimental data on mutants that are known to generate a controlled extension or collapse of protein chains. If successful, this attempt will lead to the adoption of the first force field able to simulate intrinsically disordered as well as structured proteins.
About HITS
HITS, the Heidelberg Institute for Theoretical Studies, was established in 2010 by physicist and SAP co-founder Klaus Tschira (1940-2015) and the Klaus Tschira Foundation as a private, non-profit research institute. HITS conducts basic research in the natural, mathematical, and computer sciences. Major research directions include complex simulations across scales, making sense of data, and enabling science via computational research. Application areas range from molecular biology to astrophysics. An essential characteristic of the Institute is interdisciplinarity, implemented in numerous cross-group and cross-disciplinary projects. The base funding of HITS is provided by the Klaus Tschira Foundation.